Summary
Wikipedia annotation Edit Wikipedia article
The Rfam group coordinates the annotation of Rfam families in Wikipedia. This family is described by a Wikipedia entry Flavivirus 3' UTR. More...
This page is based on a Wikipedia article. The text is available under the Creative Commons Attribution/Share-Alike License.
Sequences
Alignment
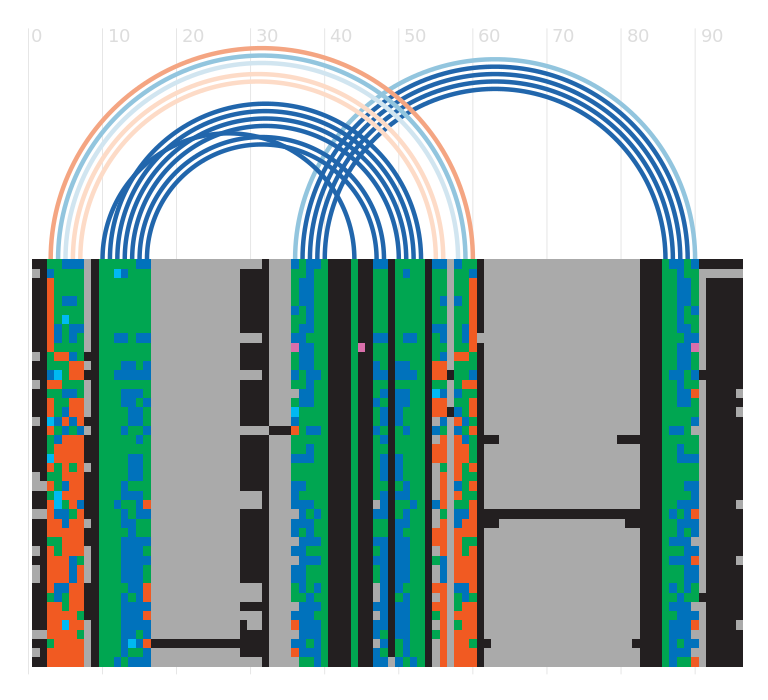
Seed alignment view
Download
Download a gzip-compressed, Stockholm-format file containing the seed alignment for this family. You may find RALEE useful when viewing sequence alignments.
Submit a new alignment
We're happy receive updated seed alignments for new or existing families. Submit your new alignment and we'll take a look.
Secondary structure
 Loading...
Loading...
Species distribution
Sunburst controls
HideWeight segments by...
Change the size of the sunburst
Colour assignments
 Archea
Archea
|
 Eukaryota
Eukaryota
|
 Bacteria
Bacteria
|
 Other sequences
Other sequences
|
 Viruses
Viruses
|
 Unclassified
Unclassified
|
 Viroids
Viroids
|
 Unclassified sequence
Unclassified sequence
|
Selections
Click on a node to select that node and its sub-tree.
Clear selection
This visualisation provides a simple graphical representation of the distribution of this family across species. You can find the original interactive tree in the adjacent tab. More...
Tree controls
HideThe tree shows the occurrence of this RNA across different species. More...
Loading...
Please note: for large trees this can take some time. While the tree is loading, you can safely switch away from this tab but if you browse away from the family page entirely, the tree will not be loaded.
Trees
This page displays the predicted phylogenetic tree for the alignment. More...
Note: You can also download the data file for the seed tree.
Structures
For those sequences which have a structure in the Protein DataBank, we generate a mapping between EMBL, PDB and Rfam coordinate systems. The table below shows the structures on which the PLRV_xrRNA family has been found.
Loading structure mapping...
References
This section shows the database cross-references that we have for this Rfam family.
Literature references
-
Steckelberg AL, Vicens Q, Costantino DA, Nix JC, Kieft JS RNA. 2020; [Epub ahead of print] The crystal structure of a Polerovirus exoribonuclease-resistant RNA shows how diverse sequences are integrated into a conserved fold. PUBMED:32848042
-
Steckelberg AL, Vicens Q, Kieft JS mBio. 2018; [Epub ahead of print] Exoribonuclease-Resistant RNAs Exist within both Coding and Noncoding Subgenomic RNAs. PUBMED:30563900
External database links
| Gene Ontology: | GO:0039694 (viral RNA genome replication); |
| Sequence Ontology: | SO:0000837 (UTR_region); |
Curation and family details
This section shows the detailed information about the Rfam family. We're happy to receive updated or improved alignments for new or existing families. Submit your new alignment and we'll take a look.
Curation
| Seed source | Vicens Q | ||||||
| Structure source | Published; PMID:32848042 | ||||||
| Type | Cis-reg; | ||||||
| Author |
Steckelberg A ,
Vicens Q,
Jeffrey K ,
Vicens Q,
Jeffrey K
|
||||||
| Alignment details |
|
Model information
| Build commands |
cmbuild -F CM SEED
cmcalibrate --mpi CM
|
| Search command |
cmsearch --cpu 4 --verbose --nohmmonly -T 25.00 -Z 2958934 CM SEQDB
|
| Gathering cutoff | 32.4 |
| Trusted cutoff | 32.4 |
| Noise cutoff | 32.3 |
| Covariance model | Download |

