Summary
Wikipedia annotation Edit Wikipedia article
The Rfam group coordinates the annotation of Rfam families in Wikipedia. This family is described by a Wikipedia entry MicroRNA. More...
This page is based on a Wikipedia article. The text is available under the Creative Commons Attribution/Share-Alike License.
Sequences
Alignment
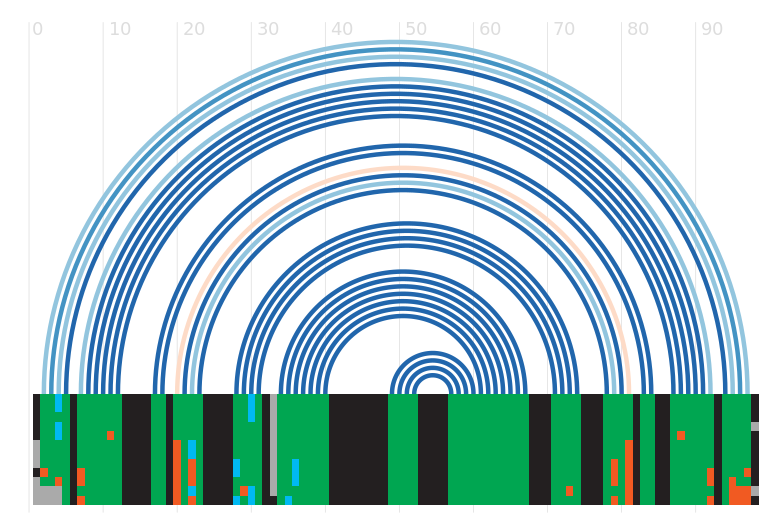
Seed alignment view
Download
Download a gzip-compressed, Stockholm-format file containing the seed alignment for this family. You may find RALEE useful when viewing sequence alignments.
Submit a new alignment
We're happy receive updated seed alignments for new or existing families. Submit your new alignment and we'll take a look.
Secondary structure
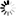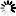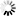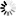 Loading...
Loading...
Species distribution
Sunburst controls
HideWeight segments by...
Change the size of the sunburst
Colour assignments
 Archea
Archea
|
 Eukaryota
Eukaryota
|
 Bacteria
Bacteria
|
 Other sequences
Other sequences
|
 Viruses
Viruses
|
 Unclassified
Unclassified
|
 Viroids
Viroids
|
 Unclassified sequence
Unclassified sequence
|
Selections
Click on a node to select that node and its sub-tree.
Clear selection
This visualisation provides a simple graphical representation of the distribution of this family across species. You can find the original interactive tree in the adjacent tab. More...
Tree controls
HideThe tree shows the occurrence of this RNA across different species. More...
Loading...
Please note: for large trees this can take some time. While the tree is loading, you can safely switch away from this tab but if you browse away from the family page entirely, the tree will not be loaded.
Trees
This page displays the predicted phylogenetic tree for the alignment. More...
Note: You can also download the data file for the seed tree.
References
This section shows the database cross-references that we have for this Rfam family.
Literature references
-
Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, Markow TA, Kaufman TC, Kellis M, Gelbart W, Iyer VN, Pollard DA, Sackton TB, Larracuente AM, Singh ND, Abad JP, Abt DN, Adryan B, Aguade M, Akashi H, Anderson WW, Aquadro CF, Ardell DH, Arguello R, Artieri CG, Barbash DA, Barker D, Barsanti P, Batterham P, Batzoglou S, Begun D, Bhutkar A, Blanco E, Bosak SA, Bradley RK, Brand AD, Brent MR, Brooks AN, Brown RH, Butlin RK, Caggese C, Calvi BR, Bernardo de Carvalho A, Caspi A, Castrezana S, Celniker SE, Chang JL, Chapple C, Chatterji S, Chinwalla A, Civetta A, Clifton SW, Comeron JM, Costello JC, Coyne JA, Daub J, David RG, Delcher AL, Delehaunty K, Do CB, Ebling H, Edwards K, Eickbush T, Evans JD, Filipski A, Findeiss S, Freyhult E, Fulton L, Fulton R, Garcia AC, Gardiner A, Garfield DA, Garvin BE, Gibson G, Gilbert D, Gnerre S, Godfrey J, Good R, Gotea V, Gravely B, Greenberg AJ, Griffiths-Jones S, Gross S, Guigo R, Gustafson EA, Haerty W, Hahn MW, Halligan DL, Halpern AL, Halter GM, Han MV, Heger A, Hillier L, Hinrichs AS, Holmes I, Hoskins RA, Hubisz MJ, Hultmark D, Huntley MA, Jaffe DB, Jagadeeshan S, Jeck WR, Johnson J, Jones CD, Jordan WC, Karpen GH, Kataoka E, Keightley PD, Kheradpour P, Kirkness EF, Koerich LB, Kristiansen K, Kudrna D, Kulathinal RJ, Kumar S, Kwok R, Lander E, Langley CH, Lapoint R, Lazzaro BP, Lee SJ, Levesque L, Li R, Lin CF, Lin MF, Lindblad-Toh K, Llopart A, Long M, Low L, Lozovsky E, Lu J, Luo M, Machado CA, Makalowski W, Marzo M, Matsuda M, Matzkin L, McAllister B, McBride CS, McKernan B, McKernan K, Mendez-Lago M, Minx P, Mollenhauer MU, Montooth K, Mount SM, Mu X, Myers E, Negre B, Newfeld S, Nielsen R, Noor MA, O'Grady P, Pachter L, Papaceit M, Parisi MJ, Parisi M, Parts L, Pedersen JS, Pesole G, Phillippy AM, Ponting CP, Pop M, Porcelli D, Powell JR, Prohaska S, Pruitt K, Puig M, Quesneville H, Ram KR, Rand D, Rasmussen MD, Reed LK, Reenan R, Reily A, Remington KA, Rieger TT, Ritchie MG, Robin C, Rogers YH, Rohde C, Rozas J, Rubenfield MJ, Ruiz A, Russo S, Salzberg SL, Sanchez-Gracia A, Saranga DJ, Sato H, Schaeffer SW, Schatz MC, Schlenke T, Schwartz R, Segarra C, Singh RS, Sirot L, Sirota M, Sisneros NB, Smith CD, Smith TF, Spieth J, Stage DE, Stark A, Stephan W, Strausberg RL, Strempel S, Sturgill D, Sutton G, Sutton GG, Tao W, Teichmann S, Tobari YN, Tomimura Y, Tsolas JM, Valente VL, Venter E, Venter JC, Vicario S, Vieira FG, Vilella AJ, Villasante A, Walenz B, Wang J, Wasserman M, Watts T, Wilson D, Wilson RK, Wing RA, Wolfner MF, Wong A, Wong GK, Wu CI, Wu G, Yamamoto D, Yang HP, Yang SP, Yorke JA, Yoshida K, Zdobnov E, Zhang P, Zhang Y, Zimin AV, Baldwin J, Abdouelleil A, Abdulkadir J, Abebe A, Abera B, Abreu J, Acer SC, Aftuck L, Alexander A, An P, Anderson E, Anderson S, Arachi H, Azer M, Bachantsang P, Barry A, Bayul T, Berlin A, Bessette D, Bloom T, Blye J, Boguslavskiy L, Bonnet C, Boukhgalter B, Bourzgui I, Brown A, Cahill P, Channer S, Cheshatsang Y, Chuda L, Citroen M, Collymore A, Cooke P, Costello M, D'Aco K, Daza R, De Haan G, DeGray S, DeMaso C, Dhargay N, Dooley K, Dooley E, Doricent M, Dorje P, Dorjee K, Dupes A, Elong R, Falk J, Farina A, Faro S, Ferguson D, Fisher S, Foley CD, Franke A, Friedrich D, Gadbois L, Gearin G, Gearin CR, Giannoukos G, Goode T, Graham J, Grandbois E, Grewal S, Gyaltsen K, Hafez N, Hagos B, Hall J, Henson C, Hollinger A, Honan T, Huard MD, Hughes L, Hurhula B, Husby ME, Kamat A, Kanga B, Kashin S, Khazanovich D, Kisner P, Lance K, Lara M, Lee W, Lennon N, Letendre F, LeVine R, Lipovsky A, Liu X, Liu J, Liu S, Lokyitsang T, Lokyitsang Y, Lubonja R, Lui A, MacDonald P, Magnisalis V, Maru K, Matthews C, McCusker W, McDonough S, Mehta T, Meldrim J, Meneus L, Mihai O, Mihalev A, Mihova T, Mittelman R, Mlenga V, Montmayeur A, Mulrain L, Navidi A, Naylor J, Negash T, Nguyen T, Nguyen N, Nicol R, Norbu C, Norbu N, Novod N, O'Neill B, Osman S, Markiewicz E, Oyono OL, Patti C, Phunkhang P, Pierre F, Priest M, Raghuraman S, Rege F, Reyes R, Rise C, Rogov P, Ross K, Ryan E, Settipalli S, Shea T, Sherpa N, Shi L, Shih D, Sparrow T, Spaulding J, Stalker J, Stange-Thomann N, Stavropoulos S, Stone C, Strader C, Tesfaye S, Thomson T, Thoulutsang Y, Thoulutsang D, Topham K, Topping I, Tsamla T, Vassiliev H, Vo A, Wangchuk T, Wangdi T, Weiand M, Wilkinson J, Wilson A, Yadav S, Young G, Yu Q, Zembek L, Zhong D, Zimmer A, Zwirko Z, Jaffe DB, Alvarez P, Brockman W, Butler J, Chin C, Gnerre S, Grabherr M, Kleber M, Mauceli E, MacCallum I Nature. 2007;450:203-218. Evolution of genes and genomes on the Drosophila phylogeny. PUBMED:17994087
External database links
| Gene Ontology: | GO:0016442 (RISC complex); GO:0035195 (miRNA-mediated post-transcriptional gene silencing); |
| Sequence Ontology: | SO:0001244 (pre_miRNA); |
| MIPF: | MIPF0000223 |
| External sites: | 1: http://www.mirbase.org |
Curation and family details
This section shows the detailed information about the Rfam family. We're happy to receive updated or improved alignments for new or existing families. Submit your new alignment and we'll take a look.
Curation
| Seed source | Griffiths-Jones SR | ||||||
| Structure source | Predicted; RNAalifold | ||||||
| Type | Gene; miRNA; | ||||||
| Author |
Griffiths-Jones SR
|
||||||
| Alignment details |
|
Model information
| Build commands |
cmbuild -F CM SEED
cmcalibrate --mpi CM
|
| Search command |
cmsearch --cpu 4 --verbose --nohmmonly -T 30.00 -Z 2958934 CM SEQDB
|
| Gathering cutoff | 60.0 |
| Trusted cutoff | 61.9 |
| Noise cutoff | 37.3 |
| Covariance model | Download |

