Summary
Wikipedia annotation Edit Wikipedia article
The Rfam group coordinates the annotation of Rfam families in Wikipedia. This family is described by a Wikipedia entry Iron response element. More...
This page is based on a Wikipedia article. The text is available under the Creative Commons Attribution/Share-Alike License.
Sequences
Alignment
Seed alignment view
Download
Download a gzip-compressed, Stockholm-format file containing the seed alignment for this family. You may find RALEE useful when viewing sequence alignments.
Submit a new alignment
We're happy receive updated seed alignments for new or existing families. Submit your new alignment and we'll take a look.
Secondary structure
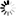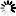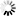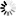 Loading...
Loading...

Species distribution
Sunburst controls
HideWeight segments by...
Change the size of the sunburst
Colour assignments
 Archea
Archea
|
 Eukaryota
Eukaryota
|
 Bacteria
Bacteria
|
 Other sequences
Other sequences
|
 Viruses
Viruses
|
 Unclassified
Unclassified
|
 Viroids
Viroids
|
 Unclassified sequence
Unclassified sequence
|
Selections
Click on a node to select that node and its sub-tree.
Clear selection
This visualisation provides a simple graphical representation of the distribution of this family across species. You can find the original interactive tree in the adjacent tab. More...
Tree controls
HideThe tree shows the occurrence of this RNA across different species. More...
Loading...
Please note: for large trees this can take some time. While the tree is loading, you can safely switch away from this tab but if you browse away from the family page entirely, the tree will not be loaded.
Trees
This page displays the predicted phylogenetic tree for the alignment. More...
Note: You can also download the data file for the seed tree.
Structures
For those sequences which have a structure in the Protein DataBank, we generate a mapping between EMBL, PDB and Rfam coordinate systems. The table below shows the structures on which the IRE_II family has been found.
Loading structure mapping...
References
This section shows the database cross-references that we have for this Rfam family.
Literature references
-
Koeller DM, Casey JL, Hentze MW, Gerhardt EM, Chan LN, Klausner RD, Harford JB Proc Natl Acad Sci U S A. 1989;86:3574-3578. A cytosolic protein binds to structural elements within the iron regulatory region of the transferrin receptor mRNA. PUBMED:2498873
-
Lymboussaki A, Pignatti E, Montosi G, Garuti C, Haile DJ, Pietrangelo A J Hepatol. 2003;39:710-715. The role of the iron responsive element in the control of ferroportin1/IREG1/MTP1 gene expression. PUBMED:14568251
-
Kohler SA, Henderson BR, Kuhn LC J Biol Chem. 1995;270:30781-30786. Succinate dehydrogenase b mRNA of Drosophila melanogaster has a functional iron-responsive element in its 5'-untranslated region. PUBMED:8530520
-
Gunshin H, Allerson CR, Polycarpou-Schwarz M, Rofts A, Rogers JT, Kishi F, Hentze MW, Rouault TA, Andrews NC, Hediger MA FEBS Lett. 2001;509:309-316. Iron-dependent regulation of the divalent metal ion transporter. PUBMED:11741608
-
Cmejla R, Petrak J, Cmejlova J Biochem Biophys Res Commun. 2006;341:158-166. A novel iron responsive element in the 3'UTR of human MRCKalpha. PUBMED:16412980
-
Gray NK, Pantopoulos K, Dandekar T, Ackrell BA, Hentze MW Proc Natl Acad Sci U S A. 1996;93:4925-4930. Translational regulation of mammalian and Drosophila citric acid cycle enzymes via iron-responsive elements. PUBMED:8643505
-
Dandekar T, Stripecke R, Gray NK, Goossen B, Constable A, Johansson HE, Hentze MW EMBO J. 1991;10:1903-1909. Identification of a novel iron-responsive element in murine and human erythroid delta-aminolevulinic acid synthase mRNA. PUBMED:2050126
-
Sanchez M, Galy B, Dandekar T, Bengert P, Vainshtein Y, Stolte J, Muckenthaler MU, Hentze MW J Biol Chem. 2006;281:22865-22874. Iron regulation and the cell cycle: identification of an iron-responsive element in the 3'-untranslated region of human cell division cycle 14A mRNA by a refined microarray-based screening strategy. PUBMED:16760464
-
Sanchez M, Galy B, Muckenthaler MU, Hentze MW Nat Struct Mol Biol. 2007;14:420-426. Iron-regulatory proteins limit hypoxia-inducible factor-2alpha expression in iron deficiency. PUBMED:17417656
-
Wingert RA, Galloway JL, Barut B, Foott H, Fraenkel P, Axe JL, Weber GJ, Dooley K, Davidson AJ, Schmid B, Paw BH, Shaw GC, Kingsley P, Palis J, Schubert H, Chen O, Kaplan J, Zon LI Nature. 2005;436:1035-1039. Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. PUBMED:16110529
-
Alen C, Sonenshein AL Proc Natl Acad Sci U S A. 1999;96:10412-10417. Bacillus subtilis aconitase is an RNA-binding protein. PUBMED:10468622
-
Kohler SA, Menotti E, Kuhn LC J Biol Chem. 1999;274:2401-2407. Molecular cloning of mouse glycolate oxidase. High evolutionary conservation and presence of an iron-responsive element-like sequence in the mRNA. PUBMED:9891009
-
Lin E, Graziano JH, Freyer GA J Biol Chem. 2001;276:27685-27692. Regulation of the 75-kDa subunit of mitochondrial complex I by iron. PUBMED:11313346
-
Walden WE, Selezneva A, Volz K FEBS Lett. 2012;586:32-35. Accommodating variety in iron-responsive elements: Crystal structure of transferrin receptor 1 B IRE bound to iron regulatory protein 1. PUBMED:22119729
External database links
| Gene Ontology: | GO:0010039 (response to iron ion); |
| Sequence Ontology: | SO:0001182 (iron_responsive_element); |
Curation and family details
This section shows the detailed information about the Rfam family. We're happy to receive updated or improved alignments for new or existing families. Submit your new alignment and we'll take a look.
Curation
| Seed source | Leipuviene, R. and Theil, E.C. | ||||||
| Structure source | Published; PMID:17849083 | ||||||
| Type | Cis-reg; | ||||||
| Author |
Stevens S,
Gardner P ,
Brown C ,
Brown C
|
||||||
| Alignment details |
|
Model information
| Build commands |
cmbuild -F CM SEED
cmcalibrate --mpi CM
|
| Search command |
cmsearch --cpu 4 --verbose --nohmmonly -T 25.00 -Z 2958934 CM SEQDB
|
| Gathering cutoff | 30.0 |
| Trusted cutoff | 30.0 |
| Noise cutoff | 29.9 |
| Covariance model | Download |

