Summary
Wikipedia annotation Edit Wikipedia article
The Rfam group coordinates the annotation of Rfam families in Wikipedia. This family is described by a Wikipedia entry Mir-193 microRNA precursor family. More...
This page is based on a Wikipedia article. The text is available under the Creative Commons Attribution/Share-Alike License.
Sequences
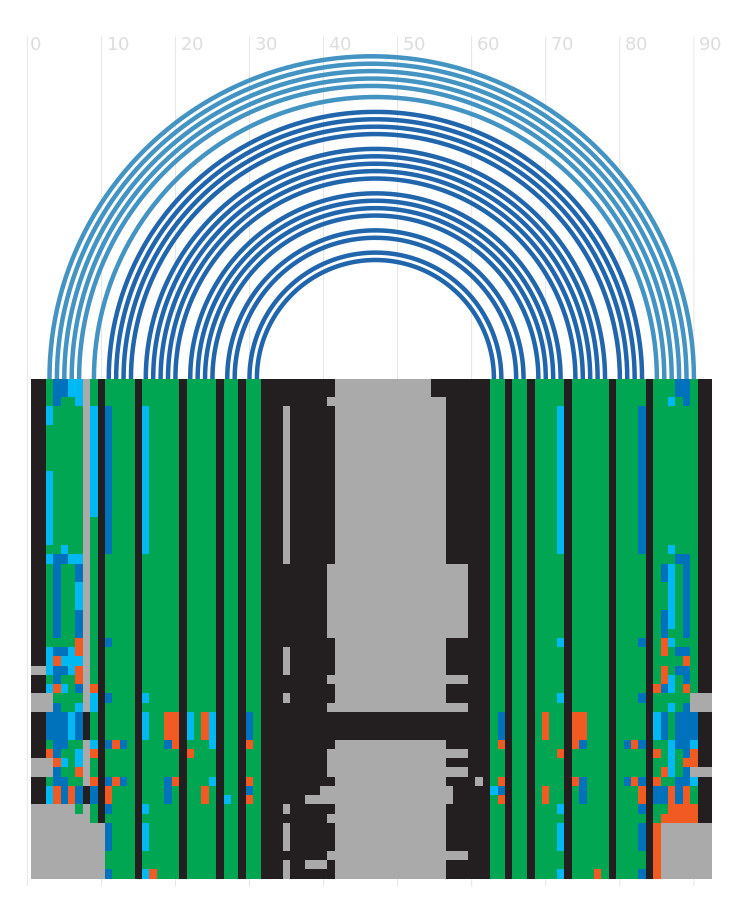
Alignment
Seed alignment view
Download
Download a gzip-compressed, Stockholm-format file containing the seed alignment for this family. You may find RALEE useful when viewing sequence alignments.
Submit a new alignment
We're happy receive updated seed alignments for new or existing families. Submit your new alignment and we'll take a look.
Secondary structure
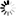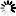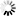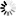 Loading...
Loading...
Species distribution
Sunburst controls
HideWeight segments by...
Change the size of the sunburst
Colour assignments
 Archea
Archea
|
 Eukaryota
Eukaryota
|
 Bacteria
Bacteria
|
 Other sequences
Other sequences
|
 Viruses
Viruses
|
 Unclassified
Unclassified
|
 Viroids
Viroids
|
 Unclassified sequence
Unclassified sequence
|
Selections
Click on a node to select that node and its sub-tree.
Clear selection
This visualisation provides a simple graphical representation of the distribution of this family across species. You can find the original interactive tree in the adjacent tab. More...
Tree controls
HideThe tree shows the occurrence of this RNA across different species. More...
Loading...
Please note: for large trees this can take some time. While the tree is loading, you can safely switch away from this tab but if you browse away from the family page entirely, the tree will not be loaded.
Trees
This page displays the predicted phylogenetic tree for the alignment. More...
Note: You can also download the data file for the seed tree.
Motif matches
There are 1 motifs which match this family.
This section shows the Rfam motifs that match sequences within the seed alignment of this family. Users should be aware that the motifs are structural constructs and do not necessarily conform to taxonomic boundaries in the way that Rfam families do. More...
| Original order | Motif Accession | Motif Description | Number of Hits | Fraction of Hits | Sum of Bits | Image |
|---|---|---|---|---|---|---|
| 7 | RM00008 | GNRA tetraloop | 2 | 0.037 | 14.8 |
|
References
This section shows the database cross-references that we have for this Rfam family.
Literature references
-
Linsen SE, de Wit E, de Bruijn E, Cuppen E; BMC Genomics. 2010;11:249. Small RNA expression and strain specificity in the rat. PUBMED:20403161
-
Lui WO, Pourmand N, Patterson BK, Fire A Cancer Res. 2007;67:6031-6043. Patterns of known and novel small RNAs in human cervical cancer. PUBMED:17616659
-
Shao P, Zhou H, Xiao ZD, He JH, Huang MB, Chen YQ, Qu LH Gene. 2008;418:34-40. Identification of novel chicken microRNAs and analysis of their genomic organization. PUBMED:18511220
-
Glazov EA, Cottee PA, Barris WC, Moore RJ, Dalrymple BP, Tizard ML Genome Res. 2008;18:957-964. A microRNA catalog of the developing chicken embryo identified by a deep sequencing approach. PUBMED:18469162
-
Cho IS, Kim J, Seo HY, Lim do H, Hong JS, Park YH, Park DC, Hong KC, Whang KY, Lee YS Mol Biol Rep. 2010;37:3567-3574. Cloning and characterization of microRNAs from porcine skeletal muscle and adipose tissue. PUBMED:20180025
-
Nielsen M, Hansen JH, Hedegaard J, Nielsen RO, Panitz F, Bendixen C, Thomsen B Anim Genet. 2010;41:159-168. MicroRNA identity and abundance in porcine skeletal muscles determined by deep sequencing. PUBMED:19917043
-
Murchison EP, Kheradpour P, Sachidanandam R, Smith C, Hodges E, Xuan Z, Kellis M, Grutzner F, Stark A, Hannon GJ Genome Res. 2008;18:995-1004. Conservation of small RNA pathways in platypus. PUBMED:18463306
-
Zhou M, Wang Q, Sun J, Li X, Xu L, Yang H, Shi H, Ning S, Chen L, Li Y, He T, Zheng Y Genomics. 2009;94:125-131. In silico detection and characteristics of novel microRNA genes in the Equus caballus genome using an integrated ab initio and comparative genomic approach PUBMED:19406225
-
Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, Sharon E, Spector Y, Bentwich Z Nat Genet. 2005;37:766-770. Identification of hundreds of conserved and nonconserved human microRNAs PUBMED:15965474
-
Strozzi F, Mazza R, Malinverni R, Williams JL Anim Genet. 2009;40:125. Annotation of 390 bovine miRNA genes by sequence similarity with other species PUBMED:18945293
-
Friedlander MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, Rajewsky N Nat Biotechnol. 2008;26:407-415. Discovering microRNAs from deep sequencing data using miRDeep PUBMED:18392026
-
Brameier M BMC Res Notes. 2010;3:64. Genome-wide comparative analysis of microRNAs in three non-human primates PUBMED:20214803
-
Ahn HW, Morin RD, Zhao H, Harris RA, Coarfa C, Chen ZJ, Milosavljevic A, Marra MA, Rajkovic A Mol Hum Reprod. 2010;16:463-471. MicroRNA transcriptome in the newborn mouse ovaries determined by massive parallel sequencing PUBMED:20215419
-
Houbaviy HB, Murray MF, Sharp PA; Dev Cell. 2003;5:351-358. Embryonic stem cell-specific MicroRNAs. PUBMED:12919684
-
Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, Lin C, Socci ND, Hermida L, Fulci V, Chiaretti S, Foa R, Schliwka J, Fuchs U, Novosel A, Muller RU, Schermer B, Bissels U, Inman J, Phan Q, Chien M, Weir DB, Choksi R, De Vita G, Frezzetti D, Trompeter HI, Hornung V, Teng G, Hartmann G, Palkovits M, Di Lauro R, Wernet P, Macino G, Rogler CE, Nagle JW, Ju J, Papavasiliou FN, Benzing T, Lichter P, Tam W, Brownstein MJ, Bosio A, Borkhardt A, Russo JJ, Sander C, Zavolan M, Tuschl T; Cell. 2007;129:1401-1414. A mammalian microRNA expression atlas based on small RNA library sequencing. PUBMED:17604727
-
Baev V, Daskalova E, Minkov I Comput Biol Chem. 2009;33:62-70. Computational identification of novel microRNA homologs in the chimpanzee genome PUBMED:18760970
-
Warren WC, Clayton DF, Ellegren H, Arnold AP, Hillier LW, Kunstner A, Searle S, White S, Vilella AJ, Fairley S, Heger A, Kong L, Ponting CP, Jarvis ED, Mello CV, Minx P, Lovell P, Velho TA, Ferris M, Balakrishnan CN, Sinha S, Blatti C, London SE, Li Y, Lin YC, George J, Sweedler J, Southey B, Gunaratne P, Watson M, Nam K, Backstrom N, Smeds L, Nabholz B, Itoh Y, Whitney O, Pfenning AR, Howard J, Volker M, Skinner BM, Griffin DK, Ye L, McLaren WM, Flicek P, Quesada V, Velasco G, Lopez-Otin C, Puente XS, Olender T, Lancet D, Smit AF, Hubley R, Konkel MK, Walker JA, Batzer MA, Gu W, Pollock DD, Chen L, Cheng Z, Eichler EE, Stapley J, Slate J, Ekblom R, Birkhead T, Burke T, Burt D, Scharff C, Adam I, Richard H, Sultan M, Soldatov A, Lehrach H, Edwards SV, Yang SP, Li X, Graves T, Fulton L, Nelson J, Chinwalla A, Hou S, Mardis ER, Wilson RK Nature. 2010;464:757-762. The genome of a songbird. PUBMED:20360741
-
Lagos-Quintana M, Rauhut R, Meyer J, Borkhardt A, Tuschl T RNA 2003;9:175-179. New microRNAs from mouse and human. PUBMED:12554859
-
Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, Johnston WK, Russ C, Luo S, Babiarz JE, Blelloch R, Schroth GP, Nusbaum C, Bartel DP Genes Dev. 2010;24:992-1009. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes PUBMED:20413612
-
Devor EJ, Samollow PB J Hered. 2008;99:66-72. In vitro and in silico annotation of conserved and nonconserved microRNAs in the genome of the marsupial Monodelphis domestica. PUBMED:17965199
-
Yue J, Sheng Y, Orwig KE BMC Genomics. 2008;9:8. Identification of novel homologous microRNA genes in the rhesus macaque genome PUBMED:18186931
-
Sewer A, Paul N, Landgraf P, Aravin A, Pfeffer S, Brownstein MJ, Tuschl T, van Nimwegen E, Zavolan M BMC Bioinformatics. 2005;6:267. Identification of clustered microRNAs using an ab initio prediction method PUBMED:16274478
-
Jin W, Grant JR, Stothard P, Moore SS, Guan LL BMC Mol Biol. 2009;10:90. Characterization of bovine miRNAs by sequencing and bioinformatics analysis PUBMED:19758457
-
Kuchenbauer F, Morin RD, Argiropoulos B, Petriv OI, Griffith M, Heuser M, Yung E, Piper J, Delaney A, Prabhu AL, Zhao Y, McDonald H, Zeng T, Hirst M, Hansen CL, Marra MA, Humphries RK Genome Res. 2008;18:1787-1797. In-depth characterization of the microRNA transcriptome in a leukemia progression model. PUBMED:18849523
-
Watanabe T, Takeda A, Tsukiyama T, Mise K, Okuno T, Sasaki H, Minami N, Imai H Genes Dev. 2006;20:1732-1743. Identification and characterization of two novel classes of small RNAs in the mouse germline: retrotransposon-derived siRNAs in oocytes and germline small RNAs in testes. PUBMED:16766679
-
Chen PY, Manninga H, Slanchev K, Chien M, Russo JJ, Ju J, Sheridan R, John B, Marks DS, Gaidatzis D, Sander C, Zavolan M, Tuschl T; Genes Dev. 2005;19:1288-1293. The developmental miRNA profiles of zebrafish as determined by small RNA cloning. PUBMED:15937218
-
Kozomara A, Birgaoanu M, Griffiths-Jones S Nucleic Acids Res. 2019;47:D155. miRBase: from microRNA sequences to function. PUBMED:30423142
External database links
| Gene Ontology: | GO:0016442 (RISC complex); GO:0035195 (miRNA-mediated post-transcriptional gene silencing); |
| Sequence Ontology: | SO:0001244 (pre_miRNA); |
| MIPF: | MIPF0000082 |
| External sites: | 1: http://www.mirbase.org |
Curation and family details
This section shows the detailed information about the Rfam family. We're happy to receive updated or improved alignments for new or existing families. Submit your new alignment and we'll take a look.
Curation
| Seed source | Griffiths-Jones SR | ||||||
| Structure source | Predicted; RNAalifold | ||||||
| Type | Gene; miRNA; | ||||||
| Author |
Griffiths-Jones SR
|
||||||
| Alignment details |
|
Model information
| Build commands |
cmbuild -F CM SEED
cmcalibrate --mpi CM
|
| Search command |
cmsearch --cpu 4 --verbose --nohmmonly -T 30.00 -Z 2958934 CM SEQDB
|
| Gathering cutoff | 55.0 |
| Trusted cutoff | 55.1 |
| Noise cutoff | 54.6 |
| Covariance model | Download |

